Mechanism of Resistance to Cancer Immunotherapy
See manuscripts in Cancer Cell (Jan 2021), Science (Jan 2018) and Nature Medicine (2018)
Many human cancers are resistant to immunotherapy for reasons that are poorly understood. We have discovered many novel resistance genes in a T cell-based genome-scale CRISPR/Cas9 screen.
See manuscripts in Cancer Cell (Jan 2021), Science (Jan 2018) and Nature Medicine (2018)
Many human cancers are resistant to immunotherapy for reasons that are poorly understood. We have discovered many novel resistance genes in a T cell-based genome-scale CRISPR/Cas9 screen.
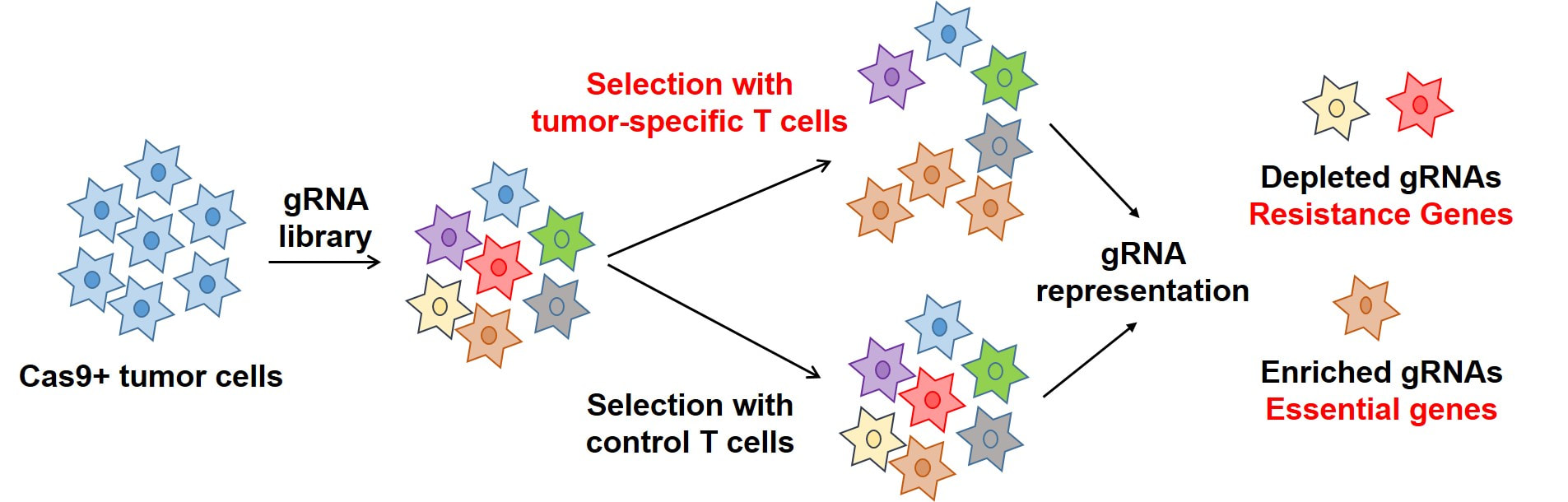
A genome-scale gRNA library is introduced into a tumor cell line that expresses Cas9. Selection is then performed with tumor-specific T cells compared to control T cells. Depleted gRNAs represent resistance genes because inactivation of these genes results in more effective T cell-mediated cytotoxicity.
Cancer immunotherapy shows limited efficacy against many solid tumors that originate from epithelial tissues, including triple-negative breast cancer (TNBC). We identified the SOX4 transcription factor as an important resistance mechanism to T cell-mediated cytotoxicity for TNBC cells.
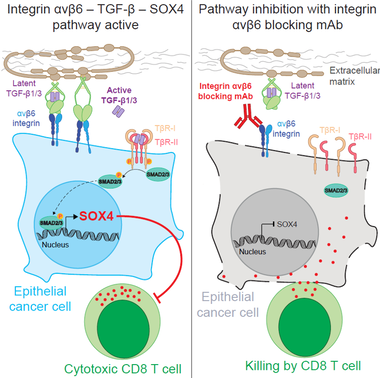
The integrin avb6 – TGFb – SOX4 pathway confers resistance to tumor cell killing by CD8 T cells.
Mechanistic studies demonstrate that inactivation of SOX4 in tumor cells increases the expression of genes in a number of innate and adaptive immune pathways important for protective tumor immunity. Expression of SOX4 is regulated by the integrin αvβ6 receptor on the surface of tumor cells, which activates TGFβ from a latent precursor. An integrin αvβ6/8-blocking monoclonal antibody (mAb) inhibits SOX4 expression and sensitizes TNBC cells to cytotoxic T cells. This integrin mAb induces a substantial survival benefit in highly metastatic murine TNBC models poorly responsive to PD-1 blockade. Targeting of the integrin αvβ6-TGFβ-SOX4 pathway therefore provides therapeutic opportunities for TNBC and other highly aggressive human cancers of epithelial origin (Cancer Cell, 2021).
We also discovered a major chromatin regulator that confers resistance, the PBAF subtype of the SWI/SNF complex. Inactivation of three genes encoding unique subunits of the PBAF complex (Pbrm1, Arid2 and Brd7) sensitized tumor cells to cytotoxic T cells (Science 2018). Loss of PBAF function increased tumor cell sensitivity to interferon-g, resulting in enhanced secretion of chemokines that recruit effector T cells into the tumor. In a back-to-back paper, clinical colleagues at Dana-Farber demonstrated that inactivating mutations of one of these genes (PBRM1) was significantly associated with better clinical responses to immunotherapy with PD-1/PD-L1 antibodies (Science 2018, 359: 801). In many human cancers, expression of PBRM1 and ARID2 inversely correlates with expression of T cell cytotoxicity genes, and Pbrm1-deficient murine melanomas are more strongly infiltrated by cytotoxic T cells. We are now characterizing other novel genes that we discovered as resistance mechanisms, with the goal of identifying novel therapeutic targets that expand the benefit of cancer immunotherapy. We are also using systematic computational approaches to discover novel resistance mechanisms, in collaboration with Dr. Shirley Liu’s lab (Nature Medicine 2018).
We also discovered a major chromatin regulator that confers resistance, the PBAF subtype of the SWI/SNF complex. Inactivation of three genes encoding unique subunits of the PBAF complex (Pbrm1, Arid2 and Brd7) sensitized tumor cells to cytotoxic T cells (Science 2018). Loss of PBAF function increased tumor cell sensitivity to interferon-g, resulting in enhanced secretion of chemokines that recruit effector T cells into the tumor. In a back-to-back paper, clinical colleagues at Dana-Farber demonstrated that inactivating mutations of one of these genes (PBRM1) was significantly associated with better clinical responses to immunotherapy with PD-1/PD-L1 antibodies (Science 2018, 359: 801). In many human cancers, expression of PBRM1 and ARID2 inversely correlates with expression of T cell cytotoxicity genes, and Pbrm1-deficient murine melanomas are more strongly infiltrated by cytotoxic T cells. We are now characterizing other novel genes that we discovered as resistance mechanisms, with the goal of identifying novel therapeutic targets that expand the benefit of cancer immunotherapy. We are also using systematic computational approaches to discover novel resistance mechanisms, in collaboration with Dr. Shirley Liu’s lab (Nature Medicine 2018).
Bagati, A., Kumar, S., Jiang, P., Pyrdol, J., Zou, A. E., Godicelj, A., Mathewson, N. D., Cartwright, A. N. R., Cejas, P., Brown, M., Giobbie-Hurder, A., Dillon, D., Agudo, J., Mittendorf, E. A., Liu, X. S., and Wucherpfennig, K. W. (2021) Integrin avb6-TGFb-SOX4 Pathway Drives Immune Evasion in Triple-Negative Breast Cancer. Cancer Cell 39, 54-67.
Pan, D., Kobayashi, A., Jiang, P., Ferrari de Andrade, L., Tay, R. E., Luoma, A. M., Tsoucas, D., Qiu, X. Lim, K., Rao, P., Long, H. W., Yuan, G. C., Doench, J., Brown, M., Liu, X. S., and Wucherpfennig, K. (2018) A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770-775
Jiang, P., Gu, S., Pan, D., Fu, J., Sahu, A., Hu, X., Li, Z., Traugh, N., Bu, X., Li, B., Liu, J., Freeman, G.J., Brown, M. A., Wucherpfennig, K. W., and Liu, X. S. (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550-1558
Pan, D., Kobayashi, A., Jiang, P., Ferrari de Andrade, L., Tay, R. E., Luoma, A. M., Tsoucas, D., Qiu, X. Lim, K., Rao, P., Long, H. W., Yuan, G. C., Doench, J., Brown, M., Liu, X. S., and Wucherpfennig, K. (2018) A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770-775
Jiang, P., Gu, S., Pan, D., Fu, J., Sahu, A., Hu, X., Li, Z., Traugh, N., Bu, X., Li, B., Liu, J., Freeman, G.J., Brown, M. A., Wucherpfennig, K. W., and Liu, X. S. (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550-1558